ARACHIDONIC acid (AA) is a polyunsaturated fatty acid mostly stored in cell membrane phospholipids in a biologically inactive, esterified form. Its free form, arachidonate, is a long-chain fatty acid anion created when AA loses a proton from its carboxy group. The amount of AA in cells is tightly regulated through the Lands cycle – a process that continuously adds and removes fatty acids from membrane phospholipids.
When needed, AA is released by phospholipase enzymes and converted into prostaglandins by COX1 and COX2 enzymes. Prostaglandins orchestrate many physiological processes, including modification of inflammatory responses. While COX1 is always present in cells, COX2 is activated by inflammatory signals and plays a key role in inflammation and cancer progression.

High levels of prostaglandins have been strongly linked to cancer progression. They help cancer cells survive and grow, increase their ability to migrate, invade other tissues, and avoid the immune response. Prostaglandins support tumor growth by directly triggering signaling pathways that control key processes like cell division, survival without attachment (anchorage-independent growth), migration, and resistance to apoptosis.
Additionally, prostaglandins, and particularly, PGE₂, influence the surrounding environment of the tumor, helping create conditions that weaken the immune response. Increased prostaglandin production is often associated with worse outcomes in lung cancer and other cancers, including gastric, liver, and colorectal cancers, as well as renal, bladder, and breast cancers.
, migration, and resistance to apoptosis.")
In many cancers – including those with KRAS mutations in lung tumors – the elevated prostaglandin levels effects have been connected to the heightened activity of COX1 and COX2, the enzymes converting arachidonic acid into prostaglandins. In lung cancer, this lipid metabolism-related mechanism is particularly relevant. Non-small cell lung cancer (NSCLC) (in this article referred to as lung cancer), which accounts for about 85% of all lung cancer cases, often involves mutations in the KRAS gene. These mutations are found in roughly 30% of NSCLC tumors. KRAS-mutant cancers tend to be more aggressive and less responsive to treatment.
The enhanced activity of COX enzymes and increased prostaglandin synthesis in such lung cancers suggest that reprogramming of AA and lipid metabolism may play a key role in supporting tumor growth and immune evasion. However, the precise mechanisms by which cancer cells alter AA metabolism to sustain high prostaglandin production remain unclear.
 can be subdivided into three main subtypes: adenocarcinoma, large cell lung carcinoma, and squamous cell carcinoma.")
Previous research shows that mutant KRAS changes how lung cancer cells use lipids. It helps cells take in extracellular fatty acids and activate them using an enzyme called acyl-CoA synthetase long-chain 3 (ACSL3). This enzyme turns long-chain fatty acids – including arachidonate – into a form the cell can use.
Because ACSL3 uses arachidonate as one of its main substrates, scientists wanted to find out if ACSL3 helps KRAS-mutant lung cancer cells make more prostaglandins. To investigate changes in lipid metabolism in this lung cancer, they used several methods, including mass spectrometry-based lipidomics analysis, cell and animal experiments, gene editing, and examination of patient tumor samples.

To investigate how exactly ACSL3 affects the KRAS-driven arachidonic acid pathway in lung cancer, researchers analyzed the lipid composition of NSCLC cells after knocking down ACSL3. Using targeted mass spectrometry, they measured the major AA-containing lipids in four phospholipid classes: phosphatidylcholine (PC), phosphatidylserine (PS), phosphatidylethanolamine (PE), and phosphatidylinositol (PI). Scientists observed that knocking down ACSL3 led to a reduction in AA content within PE and PI lipids. These findings strongly suggest that in KRAS-mutant NSCLC cells, ACSL3 helps direct AA into glycerophospholipids, especially PI. This supports the idea that ACSL3 plays a key role in attaching AA to PI in these cancer cells.
Interestingly, researchers also observed increased levels of certain PC and PS lipid species containing AA following ACSL3 knockdown in lung cancer cell lines. This suggests that when ACSL3 is reduced, the cell compensates by remodeling PC and PS phospholipids. These altered lipids could potentially be targeted by phospholipase A2 (PLA2) to release free AA for prostaglandin production.
.")
Phospholipid profile alterations after ACSL3 knockdown in lung cancer cells. Alterations in phospholipid levels following ACSL3 knockdown in a lung cancer cell line analyzed by mass spectrometry-based shotgun lipidomics. PC, phosphatidylcholine; PS, phosphatidylserine; PE, phosphatidylethanolamine; PI, phosphatidylinositol. Each group includes n = 4 samples. Results are expressed as mean ± standard deviation (SD).
Saliakoura et al., Oncogene, 39, 2948–2960 (2020), 10.1038/s41388-020-1196-5
PI is a major source of arachidonic acid, and the lipidomic analysis showed that AA-containing PI levels consistently dropped after ACSL3 knockdown in lung cancer cells. The reduction in AA-containing PIs further led to a decrease in PGE₂ production.
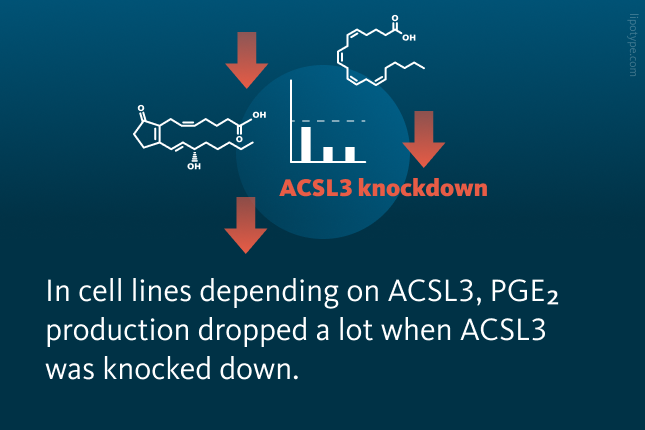
Researchers tested whether reducing ACSL3 affects PGE₂ production in lung cancer cells without KRAS mutations. In two cell lines that don’t rely on ACSL3, PGE₂ levels stayed the same or dropped slightly. In two cell lines that depend on ACSL3, PGE₂ production dropped a lot when ACSL3 was reduced. In mice with lung cancer, prostaglandin levels (like PGE₂, PGD₂, PGIPGE₂) were higher in tumors than in healthy lungs, and removing ACSL3 greatly reduced these prostaglandins in both cases. This shows that ACSL3 is important for lipid metabolism and prostaglandin production, especially in tumors where the demand for prostaglandin synthesis is higher.
.")
PGE₂ levels in the NSCLC cell lines following ACSL3 knockdown. PGE₂ levels measured by ELISA in the A549 NSCLC ACSL3 knockdown cell line. PGE₂ production was assessed 24 hours post-transduction. Each group included n = 3 samples. Data are shown as mean ± standard deviation (SD).
Saliakoura et al., Oncogene, 39, 2948–2960 (2020), 10.1038/s41388-020-1196-5
The lipid profiling in lung cancer cell culture showed that knocking down ACSL3 led to a decrease in PI 18:0/20:4, which may result from reduced conversion of LPI 18:0 into this PI species. The researchers observed a buildup of LPI 18:0, suggesting that ACSL3 knockdown limits arachidonoyl-CoA availability, thereby blocking LPI-PI conversion. The enzyme LPIAT1 is responsible for this conversion, and reducing LPIAT1 levels also led to a clear drop in PGE₂ production in KRAS-mutant NSCLC cells.
 or shRNA targeting ACSL3. Lipids were extracted after 72 hours and analyzed using mass spectrometry-based shotgun lipidomics. Each group included n = 4 samples. Data are presented as mean ± standard deviation (SD). B Schematic representation of LPIAT1’s role in phospholipid remodeling. C PGE₂ levels measured by ELISA in A549 cell line following LPIAT1 knockdown. PGE₂ production was assessed 48 hours post-transduction. Each group included n = 3 samples. Data are shown as mean ± SD. LPI, lysophosphatidylinositol; PGE₂, Prostaglandin E₂.")
LPIAT1-dependent prostaglandin synthesis requires ACSL3-derived arachidonoyl-CoA. A LPI levels measured 72 hours after ACSL3 knockdown in A549 cells. Cells were transduced with either an empty vector (control) or shRNA targeting ACSL3. Lipids were extracted after 72 hours and analyzed using mass spectrometry-based shotgun lipidomics. Each group included n = 4 samples. Data are presented as mean ± standard deviation (SD). B Schematic representation of LPIAT1’s role in phospholipid remodeling. C PGE₂ levels measured by ELISA in A549 cell line following LPIAT1 knockdown. PGE₂ production was assessed 48 hours post-transduction. Each group included n = 3 samples. Data are shown as mean ± SD. LPI, lysophosphatidylinositol; PGE₂, Prostaglandin E₂.
Saliakoura et al., Oncogene, 39, 2948–2960 (2020), 10.1038/s41388-020-1196-5
Researchers tested whether reducing LPIAT1 affects PGE₂ production and cell growth in lung cancer cells with normal KRAS. In cells not dependent on ACSL3, LPIAT1 knockdown had little or no effect. However, in ACSL3-dependent cells, it lowered PGE₂ levels and slowed growth. It also reduced growth in several KRAS-mutant cell lines. This suggests that ACSL3 and LPIAT1 work together to support prostaglandin production and cancer cell growth, and LPIAT1 needs ACSL3 to support cancer cell growth and prostaglandin production. Knocking down LPIAT1 in mice with human lung tumors suppressed tumor growth and improved survival. In human NSCLC, LPIAT1 is often higher and linked to worse outcomes. Researchers found that LPIAT1 and ACSL3 levels are closely linked and that high levels of either are associated with lower patient survival.
Overall, scientists identified the ACSL3–LPIAT1 metabolic axis as a key regulator of prostaglandin production and cancer cell proliferation in non-small-cell lung cancer. Cells sensitive to ACSL3 knockdown are also sensitive to LPIAT1 knockdown, suggesting they act in the same lipid metabolism pathway. Overexpression of LPIAT1 did promote proliferation, which was blocked by ACSL3 knockdown, likely by limiting arachidonoyl-CoA availability. While some cell lines resist this inhibition, indicating alternative pathways, targeting ACSL3–LPIAT1 may offer a tumor-specific therapeutic strategy with fewer side effects than COX inhibitors.




Lipotype Lipidomics technology performs detailed analysis of lipid composition in biological systems. This helps to evaluate how specific lipids, such as arachidonic acid and other fatty acids, contribute to disease processes like cancer, and supports the development of targeted therapies, biomarkers, and drug delivery strategies.
Need clarity on the process?
Ask us anything!

Lipotype products are provided for Research Use Only. They are not intended for clinical diagnostic purposes and must not be used to inform medical treatment decisions. The content of this article is for scientific and educational purposes only and should not be considered medical advice.